Getting Started with Graph Neural Networks for Protein–Ligand Complexes Using DGL
A practical, beginner-friendly introduction to the Deep Graph Library (DGL) and how to use it to featurize protein–ligand complexes for machine learning in drug discovery.
Apr 5, 2026
What Agentic Engineering Means for Computational Drug Discovery
Simon Willison recently appeared on Lenny’s Podcast to discuss what he calls the November inflection point: the moment in late 2025 when frontier models crossed a threshold where agentic coding went from “mostly works if you watch carefully” to “almost always does what you asked.” His highlights post is worth reading in full, but reading it through the lens of computational drug discovery, several themes land with unusual force.
Apr 4, 2026
Beyond 2D Fingerprints: Encoding Protein-Ligand Interactions for Machine Learning
A practical guide to three advanced 3D fingerprinting methods (PLEC, SPLIF, and E3FP) and how to choose between them when featurizing docking poses for ML-based drug discovery models.
Apr 1, 2026
Understanding Binding Energetics in Molecular Docking
Conceptual overview of the key energetic contributions governing protein–ligand binding in molecular docking, including desolvation, entropy, water displacement, electrostatics, and scoring function behavior.
Mar 13, 2026
Practical System Preparation Tips for Molecular Dynamics Simulations
Preparing a molecular system correctly before running molecular dynamics (MD) simulations is essential for obtaining meaningful and reproducible results. Small technical choices such as solvent box geometry, treatment of protein termini, and strategies for selecting representative conformations can strongly influence simulation stability, computational efficiency, and interpretation of results.
Mar 9, 2026
Unlocking the Undruggable: How Biotechnology Is Rewriting Drug Discovery
A concise overview of how modern biotechnology, new therapeutic modalities, and smarter development strategies are transforming previously 'undruggable' targets into tractable opportunities.
Nov 22, 2025
How to Use DataWarrior for Drug Discovery: Key Workflows From the Villoutreix Tutorials
Practical introduction to DataWarrior as a free, chemistry-aware workbench for data visualization, filtering, and focused library generation, based on Bruno Villoutreix’s tutorial series.
Nov 15, 2025
Energy Minimization with Open Babel: Practical Guide for Ligand Preparation
Step-by-step workflow for energy minimization of ligands using Open Babel’s obminimize tool, with comparisons of force fields and minimization algorithms for docking and molecular modeling.
Sep 23, 2025
Awesome Drug Discovery
Aug 25, 2025
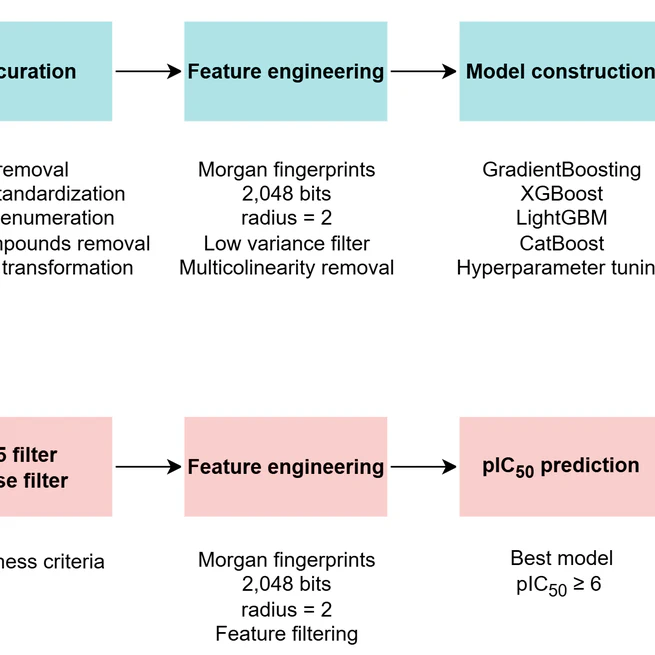
QSARBoost
Aug 15, 2025