Understanding Molecular Dynamics Simulations
Molecular Dynamics (MD) simulation is a computational technique for studying the behavior of complex molecular systems at atomic resolution. MD works by constructing an initial configuration of a system, including the positions, velocities, and masses of all atoms or molecules. The simulation calculates the forces acting on each particle from its interactions with neighbors.
These forces update the positions and velocities of each particle over a small time step, typically in the femtosecond to picosecond range. Repeating this process over many steps allows the system to evolve over time.
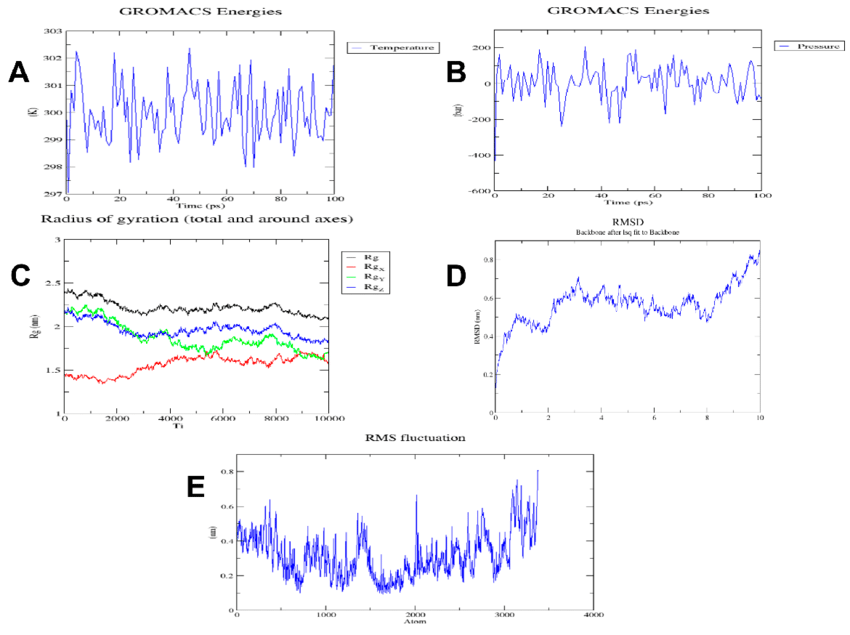 |
|---|
| Figure 1: Example MD simulation output plots |
MD simulation provides insights into molecular systems that are difficult or impossible to study experimentally. In structural biology, it is used to study protein dynamics, including conformational changes, ligand binding, and intermolecular interactions. By simulating atomic-level motion, researchers can connect protein structure to function in ways that static experimental structures cannot.
MD simulation is also widely used in the field of materials science. It can model the behavior of polymers, ceramics, metals, and other materials, providing insight into their mechanical properties, stability, and behavior under different conditions. By simulating the behavior of materials at the atomic scale, researchers can develop a deeper understanding of how they behave and how they can be optimized for specific applications.
A key strength of MD is its ability to handle systems with large numbers of particles. Proteins, which contain thousands of atoms, are difficult to characterize experimentally at this resolution. MD allows detailed study of such systems, including structure, dynamics, and function.
Another advantage of MD simulation is its ability to simulate systems under a wide range of conditions, including different temperatures, pressures, and chemical environments. This makes it a powerful tool for studying how systems behave under different conditions and how they respond to changes in their environment.
While MD simulation has its limitations, such as accurately modeling the interactions between particles and the computational cost, advances in computing technology have made it more accessible. Researchers are constantly developing new algorithms and techniques to improve the accuracy and efficiency of simulations.
Common outputs from MD simulations include trajectories (particle positions over time), energy plots, radial distribution functions, and density profiles. Together, these give a quantitative picture of system stability, solvation, and structural evolution.
MD simulation is broadly applicable across structural biology, materials science, and computational drug discovery. As hardware and algorithms continue to improve, it remains one of the most informative tools for studying molecular behavior at atomistic resolution.