Principal Component Analysis and Free Energy Landscape Mapping Using GROMACS
Step-by-step workflow for performing PCA and Free Energy Landscape (FEL) analysis from MD simulations with GROMACS.
Jul 9, 2025
Validating Molecular Docking Poses with DFT: A Quick Guide
Learn how to use Density Functional Theory (DFT) to validate docking poses, estimate strain energies, and gain quantum insights into ligand binding.
Jul 3, 2025
Step-by-Step MD Simulation of a Protein–Ligand Complex with GROMACS
A clear workflow for setting up, running, and analyzing a protein–ligand complex simulation using CHARMM36 and TIP3P in GROMACS.
Apr 4, 2025
Molecular Simulation in Drug Discovery: A Strategic Guide to Core Methods
Overview of key molecular simulation methods (QM, MD, CGMD, MC, BD, LD, DPD) and their strategic applications in drug discovery pipelines.
Jan 8, 2025
How to Validate AlphaFold Structures
Practical guide to checking AlphaFold-predicted protein models using pLDDT, PAE, geometry validation tools, structural comparisons, and functional tests.
Mar 28, 2024
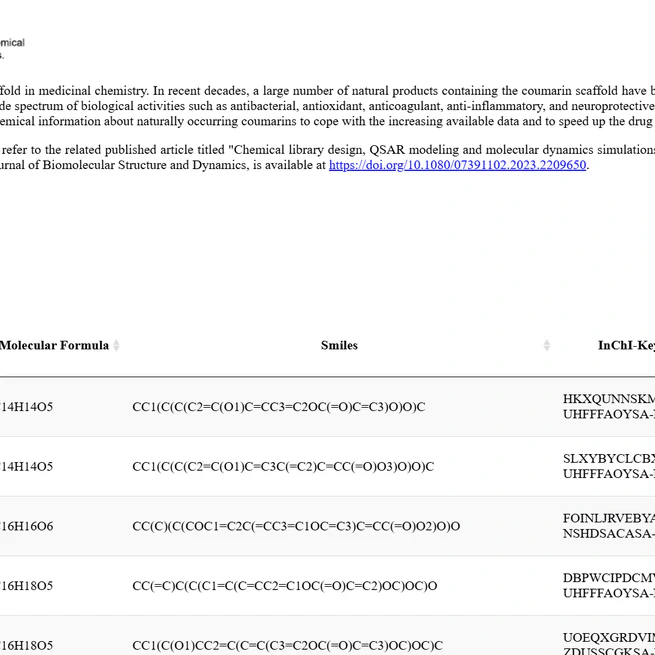
CoumarinDB
Jan 25, 2024
How to build and validate 3D-QSAR models - Insights from Xu et al., 2020
Comprehensive, metric-driven workflow for robust 3D-QSAR modeling, based on Xu et al., 2020 and enriched with cheminformatics best practices.
Jan 13, 2024
How Long Should Molecular Dynamics Simulations Run? A Practical Guide
Guidelines for choosing appropriate MD simulation lengths in protein–ligand systems, with key convergence metrics like RMSD, RMSF, and interaction fingerprints.
Jan 9, 2023