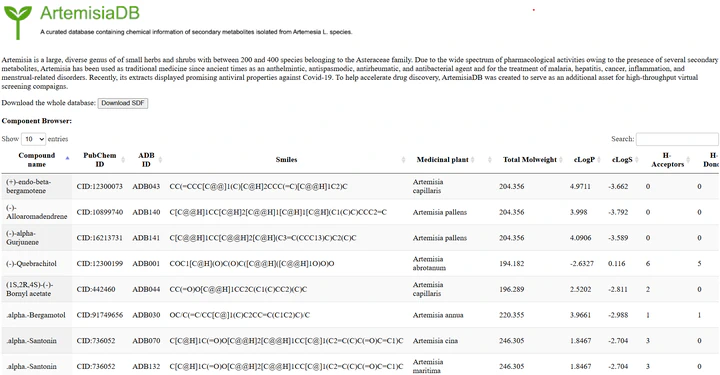
ArtemisiaDB Jan 25, 2024 · 0 min read Go to Project Site Last updated on Jan 25, 2024 Database Natural Products Virtual Screening Artemisia Authors Yassir Boulaamane Postdoctoral Researcher ← EnsembleBBB Feb 19, 2024 CoumarinDB Jan 25, 2024 →